Historically, quantifying DNA has been a useful tool to learn more about cellular functions, and quantify at what stage in the cell cycle the cell is. Calculating the amount of DNA present was usually performed by binding a fluorophore (fluorescent chemical compound that can re-emit light upon light excitation) in a specific ratio to the cell’s DNA, and measuring changes through flow cytometry in a high throughput fashion (calculating the change of DNA matter from a large number of cells).
While this method allowed for the study of certain aspects of cell populations, shifting dynamics of a cellular population (as well as variables within populations of diverse cells) did not allow for examination of a cell on the individual level. Additionally, exposing a delicate and unstable cell population to toxins that influence behavior would result in an inaccurate understanding of true cellular content and behavior.
Previously, visualization studies have been performed by incorporating highly specific laser scanning cytometry and fluorescent markers. Many of the fluorophores used were (and are) incompatible with live cells (for example, DAPI), since they are not membrane permeable chemicals, and force the researcher to have the cells fixed through chemical means (and thus frozen in time) before the cell can be labeled. Membrane permeable fluorophores have recently been designed to visualize the movement of live chromosomes over time, but it quickly became evident that some of the most common dyes contained cytotoxins that initiated DNA mutation and trigger certain genes (such as p53) to become activated. Additionally, the UV light exposure needed for the experiment would eventually cause DNA to break apart, leading to cell cycle arrest and apoptosis.
Clearly, the previously mentioned methodologies to study cellular behavior (particularly the behavior of unstable cancer cells) cannot be used when the reagents influence the outcome of the experiment, so new methods were necessary.
Gomes et al at the University of Arizona Cancer Center have recently published a methodology paper in Cell Division where they attempted to bridge the gap between cell visualization and unaltered cellular environment by using a non-toxic procedure to measure DNA content in live cells over a period of time. They used the fluorophore Hoechst 33342, a live-cell DNA dye that is permeable to the cellular membrane, binds AT regions of the DNA strand, and is compatible with living cells in a stable environment (it should not induce cytotoxic effects that interfere with the study of the cell). This methodology also allows for the use of non-3D imaging, making the entire process easier and faster for scientists.
Gomes demonstrated through these experiments that is it possible to monitor chromosomal dynamics in cells (altered to express a GFP-histone 2B fusion protein) of varying ploidies through the following steps:
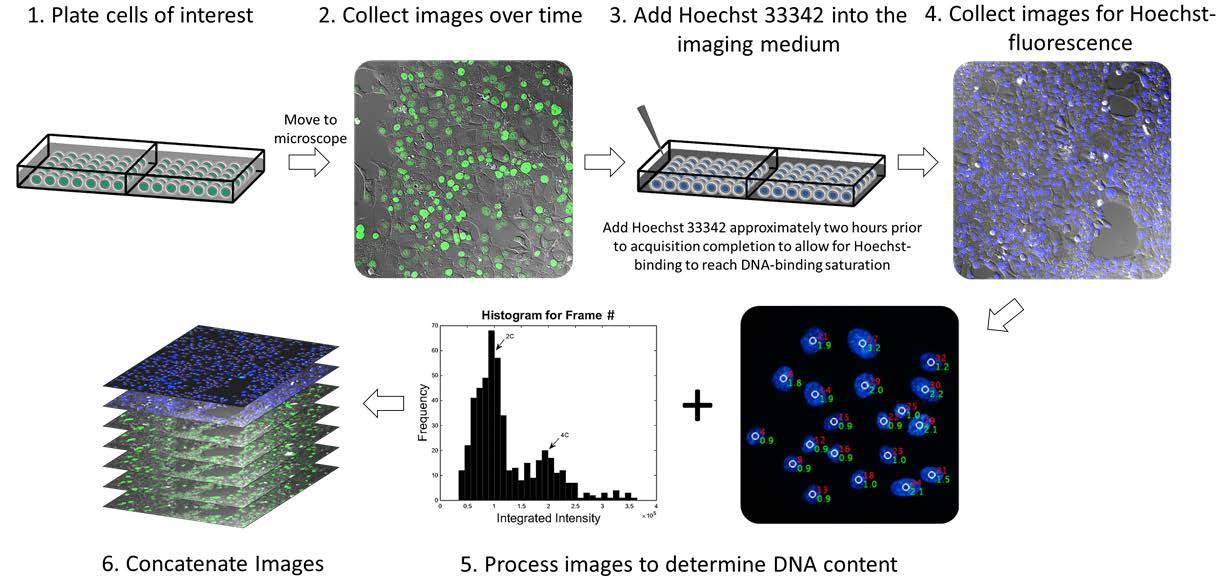
This new methodology allows scientists to visualize cells that are highly genetically unstable (such as cancer cells) without interfering with genes that are thought to be involved in the mutations that lead to cancer.
Of particular note, it was possible to visualize the following error prone mitoses:
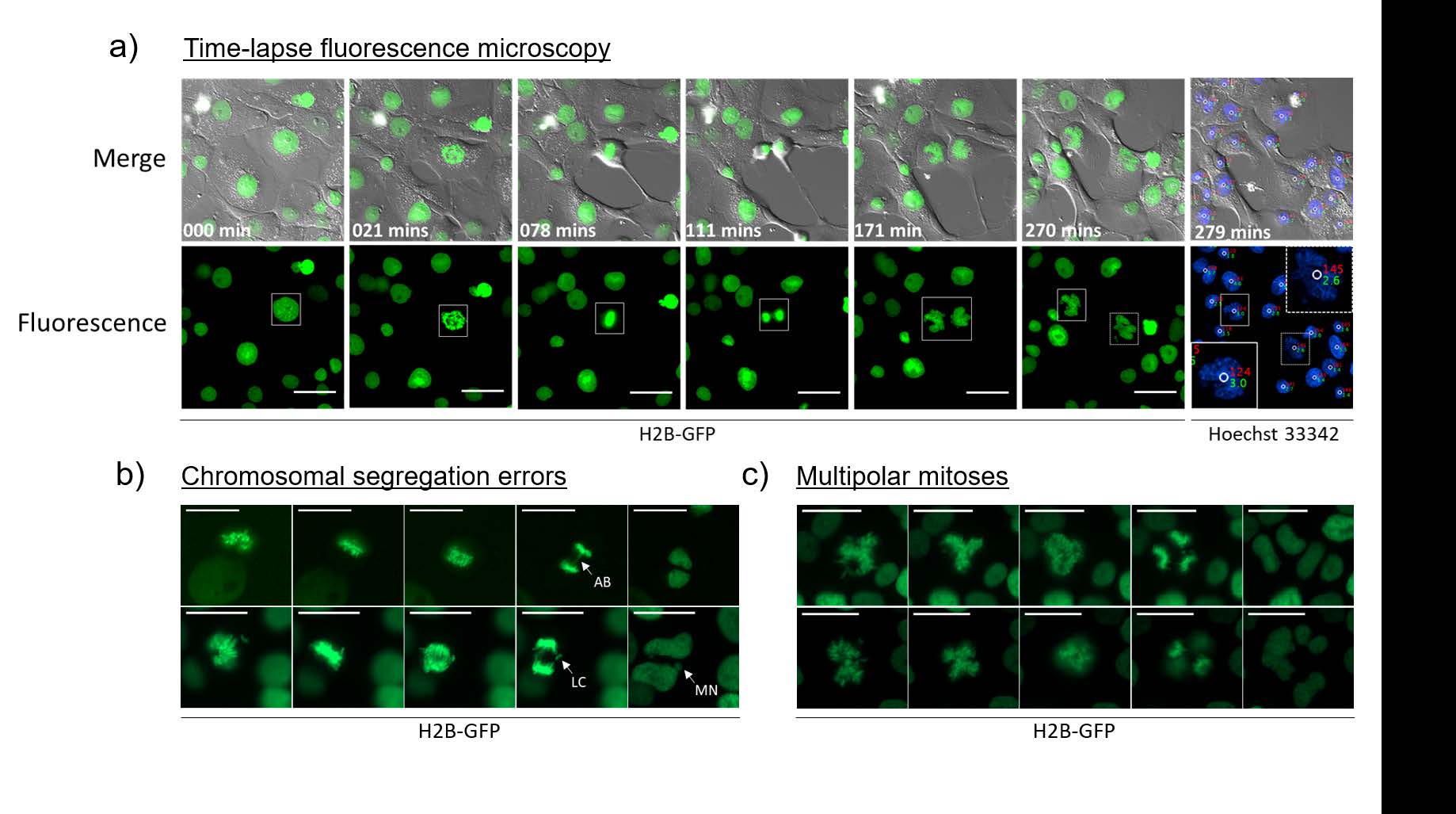
This breakthrough in imaging methodology provides scientists the tools to watch cancer cells grow and mutate, in ways that were previously cumbersome and difficult to confirm. It is an attractive new technique for exploring unresolved cell division and proliferation dynamics of polyploid cells, without the need for expensive and complicated 3D imaging, and according to the authors can be used with any stoichiometric live-cell DNA dye. Beyond cancer research, this imaging modality provides researchers of various disciplines the improved ability to learn and understand more about cellular behavior and processes.
**Original video and all images taken from Measuring DNA content in live cells by fluorescence microscopy by Gomes et. al. at the University of Arizona.
Faye Nourollahi
Latest posts by Faye Nourollahi (see all)
- Watching cancer cells take over - 8th October 2018
- 3rd International Day of Women and Girls in Science at the United Nations - 12th February 2018
Comments