The Cambridge physiologist John Newport Langley proposed in 1909 the presence of a “receptive substance” that mediated drug action. This idea remained hypothetical until two further breakthroughs – when Raymond P. Ahlquist proposed in 1948 the existence of two receivers for epinephrine (α- and β-adrenoceptors), and when drugs able to block such receivers were developed in 1965. Blockers were and still are instrumental to combat diseases. One of the first was propranolol, the first clinically useful β-adrenoceptor blocker, for which James Black was awarded the Nobel Prize in 1988.
We now know that GPCRs are highly diversified in mammalian genomes (with humans estimated to have about one thousand GPCR genes – 8% of the human proteome!)
After the discovery by Alfred G. Gilman (Nobel Prize in 1994) of G proteins, Langley’s “receptive substances” were identified as the G-protein-coupled receptor (GPCR) family of proteins. G proteins are found on the inner surface of the cell membrane and propagate into the cell a signal initiated by the binding of a hormone –for instance, epinephrine – to one of its cognate GPCRs on the cell surface.
Molecular biology techniques have helped to identify amino acid sequences, and significant advances in crystallization techniques have helped solve the crystal structures of dozens of GPCRs, establishing their archetypal monomeric structure as a seven-transmembrane domain protein coupled to a trimeric G protein. The importance of GPCR research was recognized in 2012, when the Nobel Prize was awarded to Robert J. Lefkowitz and Brian K. Kobilka, for their work on this family of proteins.
We now know that GPCRs are highly diversified in mammalian genomes (with humans estimated to have about one thousand GPCR genes – 8% of the human proteome!), and they are the target of about 30% of the drugs sold in Pharmacies. Despite all the significant advances, however, their role in mediating drug action and signal transduction into the cell is still not fully understood.
A GPCR quartet as a signaling switch
We are interested in the signaling function of GPCRs, and in particular in the receptors that respond to adenosine, one of the most ancient hormones. Adenosine receptors regulate a broad range of bodily functions, including energy and temperature homeostasis and neurotransmitter release. But interestingly, they also mediate the effects exerted by caffeine in coffee and cola drinks, theophylline in tea, and theobromine in cacao.
As with many other GPCRs, adenosine receptors in mammals are a subfamily of related proteins (A1, A2A, A2B and A3). These may form, in addition to functional singular units (monomers), higher-order complexes (oligomers) comprised of a number of equal (homo) or different (hetero) monomers. Hence, we thought of performing research to understand the reason why GPCR dimers/oligomers exist in nature.
In two contributions published in BMC Biology, we have now shown that the minimal unit of the adenosine sensor device is constituted by four receptors, two A1R and two A2AR, simultaneously coupled to two G proteins, one Gi and one Gs.
The origin of the current discovery started 12 years ago after a serendipitous finding. With the aim of showing a lack of interaction between A1R, which is coupled to the Gi protein (mediating the inhibition of adenylate cyclase and decrease of cAMP), and A2AR, which is coupled to Gs (mediating the activation of adenylate cyclase and increase of cAMP), we found the opposite. A1R and A2AR do form heteromers, with a signaling output that cannot be accounted for as the sum of signals from its constituents signaling. This confers A1R-A2AR complexes the property of being able to act as sensitive sensors of the concentration of adenosine, both in cells in culture and in nerve terminals in the brain. However, the mechanism through which this is achieved was unclear.
In two contributions published in BMC Biology, we have now shown that the minimal unit of the adenosine sensor device is constituted by four receptors, two A1R and two A2AR, simultaneously coupled to two G proteins, one Gi and one Gs. Combining experiments with computational models we proposed the overall structure consisting of a compact rhombus-shaped heterotetramer.
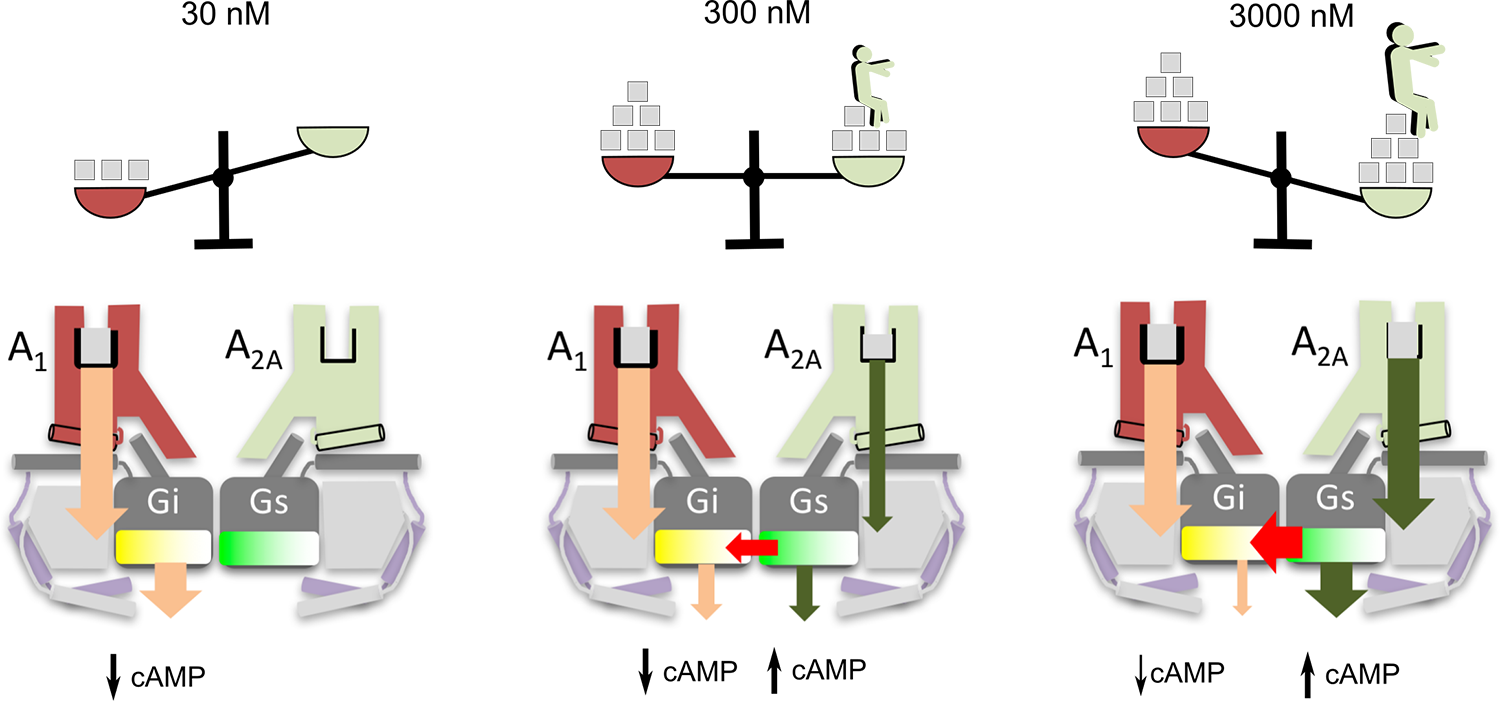
In the second contribution, we have shown the mechanism by which the sensor works at the molecular level. Due to A1R’s higher affinity for the hormone, adenosine at a low concentration binds predominantly to A1R, engaging Gi-mediated signaling, which significantly decreases cAMP accumulation (left panel). However, at higher concentrations, adenosine progressively binds to A2AR (middle panel), until at saturating concentrations, with full occupancy of both A1R and A2AR, there is a marked increase in cAMP levels because Gs is activated while Gi is blocked. Our structural model suggests that in the compact tetrameric complex, the C-terminal tail of the activated A2A receptor prevents the simultaneous activation of both Gs and Gi, leading instead to preferential Gs coupling.
A challenge for the 21st century is to understand how other GPCR (hetero)oligomers influence the physiological role of receptors, and to assess their potential as therapeutic targets. The several hundred GPCRs in higher organisms already provide diversity but we now need to also consider what are the novel properties arising when GPCRs form heteromers. The number of identified heteromers is now counted by hundreds, and they include receptors for peptides and photons as well as hormones. Clearly, the possibility of targeting or disrupting physiologically relevant GPCR heteromers provides new opportunities for novel drug discovery.
Comments