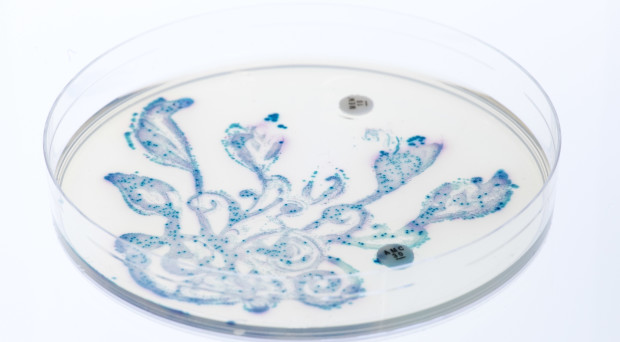
The advances of metagenomics have given us the possibility to characterise the bacterial composition in samples of their habitats, including the human body. Such identification sheds further light on the crucial human-microbe symbiosis that has a profound impacton human health and disease development. A next, necessary step, is to map and understand the interactions between different bacterial communities themselves. For this purpose, Mitch Fernandez and collaborators show how the outcome of sequencing and classification analyses can be used to generate bacterial co-occurrence networks and within these, through Markov clustering approaches, identify “clubs”, “club leaders” and “rival clubs”. As examples, the authors apply their methods on two datasets: analysis of microbes in airways of smokers and former smokers suggests that the bacterial communities of active smokers might experience greater rivalry among themselves compared to those in former smokers; while investigation of data from the Human Microbiome Project show how the number of clubs and their interplay differ with the locations in the human body.

Assessing structural variation in a personal genome—towards a human reference diploid genome
Full genome sequencing becomes increasingly more feasible, enabling more detailed and complex analysis of the genome. One topic of particular interest concerns structural variants of the chromosomes, encompassing copy-number variants, inversions, mobile element insertions, deletions and translocations, which are of importance for understanding the genetic and phenotypic diversity, and in some cases disease development. Such structural variants are however more challenging to detect compared to single-nucleotide variants and smaller indels. Current methods are often restricted to handle certain types, sizes or locations. This is why Adam English and collaborators present ‘Parliament’, a tool that merges and analyses data from an ensemble of structural variation discovery programs. The authors illustrate this infrastructure with the genome of one human individual, integrating paired-end, array comparative genomic hybridization, long-read, long-insert, and whole-genome architecture data. 9,777 structural variations are identified, spanning at least 1.8% of the reference genome. With this, the researchers hope to have introduced a user-friendly analysis tool, as well as contributed towards a reference diploid genome that could serve as a gold standard description of structural variation.
As if taken from a horror film, the fungi Ophiocordyceps unilateralis are parasites which, when infecting a Carpenter ant’s brain, change the host’s behaviour and causes it to leave the nest at a different time compared to the rest of the ants, walk aberrantly, and ultimately bite onto a plant with a lock-jaw that ensures that the ant remains there even after its death. While outwards seeming erratic, these manipulated behaviours provide environmental conditions fitting the fungal development and life cycle. The control of the ant is assumed to be induced through the gene expression of the fungus, but the process has not yet been fully examined. In a previous study, discussed in an earlier blog, Charissa de Bekker and collaborators investigated how the parasite affects its natural host compared to novel ants, and identified a set of molecules that the fungi release when in contact with ant brains.
Here the researchers in a laboratory environment set out to infect Carpenter ants, and sequence sample tissues from the host and the parasite collected when manipulated biting was observed. In this hitherto uncharted O. unilateralis s.l. genome, the authors find dynamically expressed genes encoding for enterotoxins that might affect the chemosignalling molecules and up-regulate apoptotic processes in the host. Ant genes possibly associated with immune and stress responses are down-regulated, while clock gene homologues and genes involved in dopamine metabolism are up-regulated. The fungus might further steer the infected ant through up-regulation of genes related to production of compounds which can interfere with the receptors of the host nervous system, alter the neuronal cell membrane, or shift metabolic pathways.

Mobile element insertions are frequent in oesophageal adenocarcinomas and can mislead paired-end sequencing analysis
While taking part in the ‘Int ernational Cancer Genome Consortium framework programme’ of sequencing 500 oesophageal adenocarcinomas, Anna Paterson and her colleagues from University of Cambridge decided to investigate a puzzling type of genome rearrangement that had also been noted by other researchers. Paired-end sequence analysis data seemed to suggest that a number of tumours underwent unorthodox chromosome translocation events, the same observations often being made for several tumour samples.
ernational Cancer Genome Consortium framework programme’ of sequencing 500 oesophageal adenocarcinomas, Anna Paterson and her colleagues from University of Cambridge decided to investigate a puzzling type of genome rearrangement that had also been noted by other researchers. Paired-end sequence analysis data seemed to suggest that a number of tumours underwent unorthodox chromosome translocation events, the same observations often being made for several tumour samples.
Examining this enigma closer, the authors showed that the observed rearrangements in reality are insertions of type L1 mobile elements. Apart from illustrating that such inserts may be problematic for interpreting outcomes obtained with current paired-end sequencing procedures, these results also demonstrate that mobile insertions may in fact occur in the majority of oesophageal adenocarcinomas. The possibility that they might affect the activation of genes could be another route towards causing cellular disruption which ultimately leads to cancer.
Comments